
Unveiling the Chemical Reactions Involved in Moisture-Induced Weakening of Adhesion between Aluminum and Epoxy Resin
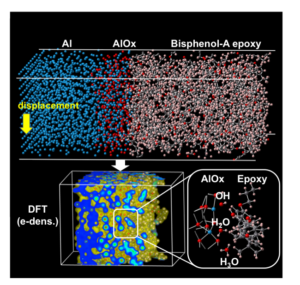 Epoxy resin is commonly used as an adhesive for bonding metals. Recent industrial demands, for example, lightweight multimaterial cars and highly reliable pressure sensors, motivate researchers to improve the reliability and durability of bonded materials. One critical issue regarding adhesive bonding is the weakening of its bonding strength by the water that migrates from a moist environment along the interface. To understand the mechanisms at the electronic structure level, we perform atomic dynamics simulations on various Al and epoxy resin interface systems with water molecules inserted in the contact region. In accordance with experimental conditions, the Al layer is surface-oxidized while the bisphenol-A type epoxy molecule has both OH and ether groups. Shear deformations are simulated using the hybrid quantum-classical method in which about 1500 atoms at the contact region are treated with the density-functional theory using out DC-RGDFT code. For the first time, calculated adhesion strengths compare well with the experimental values. Three types of chemical reactions that affect the adhesion strength occur depending on the terminal functional groups of the Al oxide surface and the water layer formation. Separate calculations confirm small barrier energies for all of the reaction processes.
Epoxy resin is commonly used as an adhesive for bonding metals. Recent industrial demands, for example, lightweight multimaterial cars and highly reliable pressure sensors, motivate researchers to improve the reliability and durability of bonded materials. One critical issue regarding adhesive bonding is the weakening of its bonding strength by the water that migrates from a moist environment along the interface. To understand the mechanisms at the electronic structure level, we perform atomic dynamics simulations on various Al and epoxy resin interface systems with water molecules inserted in the contact region. In accordance with experimental conditions, the Al layer is surface-oxidized while the bisphenol-A type epoxy molecule has both OH and ether groups. Shear deformations are simulated using the hybrid quantum-classical method in which about 1500 atoms at the contact region are treated with the density-functional theory using out DC-RGDFT code. For the first time, calculated adhesion strengths compare well with the experimental values. Three types of chemical reactions that affect the adhesion strength occur depending on the terminal functional groups of the Al oxide surface and the water layer formation. Separate calculations confirm small barrier energies for all of the reaction processes.
Key words: Organic-inorganic interface, epoxy resin, Al, adhesion, hybrid simulation
Development of electronic state calculation code of large scale system
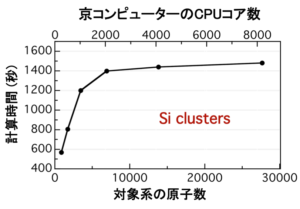 We are researching and developing new electronic state calculation method and its general purpose code which can be applied to large scale target system of thousands of atomic scale. The code of the density functional method based on the Kohn-Sham equation is often expressed and calculated by simply expressing the electron orbit using many plane waves. However, as a result, efficiency is not good when calculating with many nodes of supercomputer. Therefore, in the simulation to investigate the dynamics of atoms, the number of atoms is on the order of 100 in practical use. On the other hand, we developed a real space grid density functional code (RGDFT) that expresses orbital functions as numerical values on lattice points in real space. Since it is a real space expression, boundary condition and total charge number can be freely set. In this method, high latency performance can be obtained when many nodes of supercomputer are used by virtually dividing all lattice points to reduce the number of lattice points each node is in charge of. Recently, in order to be able to deal with the target system of thousands of atomic size, the target system itself is also virtually divided into domains, and a new formulation that integrates the results of real space grid density functional calculations in each domain is also proposed, (DC-RGDFT). According to this method, the force acting on all atoms of the target system can be obtained with a calculation amount proportional to the number of atoms. DC-RGDFT is a performance test using a number of nodes in Supercomputer and shows good results.
We are researching and developing new electronic state calculation method and its general purpose code which can be applied to large scale target system of thousands of atomic scale. The code of the density functional method based on the Kohn-Sham equation is often expressed and calculated by simply expressing the electron orbit using many plane waves. However, as a result, efficiency is not good when calculating with many nodes of supercomputer. Therefore, in the simulation to investigate the dynamics of atoms, the number of atoms is on the order of 100 in practical use. On the other hand, we developed a real space grid density functional code (RGDFT) that expresses orbital functions as numerical values on lattice points in real space. Since it is a real space expression, boundary condition and total charge number can be freely set. In this method, high latency performance can be obtained when many nodes of supercomputer are used by virtually dividing all lattice points to reduce the number of lattice points each node is in charge of. Recently, in order to be able to deal with the target system of thousands of atomic size, the target system itself is also virtually divided into domains, and a new formulation that integrates the results of real space grid density functional calculations in each domain is also proposed, (DC-RGDFT). According to this method, the force acting on all atoms of the target system can be obtained with a calculation amount proportional to the number of atoms. DC-RGDFT is a performance test using a number of nodes in Supercomputer and shows good results.
Key words: quantum mechanics, first principles calculation, supercomputer
First principle simulation of negative electrode SEI of Li ion secondary battery
 Li ion batteries are currently most used mainly for electronic devices, because of its high voltage among various types of storage batteries, capable of repeated use many times and being compact. However, in order to use Li ion batteries as power for automobiles and the like, it is necessary to further improve the performance in various aspects. In the interior of the Li-ion battery, a thin film called a solid electrolyte film (SEI) is formed at the interface between the negative electrode and the electrolyte at the initial charge. Increasing the rate at which Li ions pass through the SEI is important for improving the output performance of the battery. We used the electron state calculation code DC-RGDFT developed independently to calculate 5 times the Li ion group passing through the SEI – electrolyte interface at the vicinity of the Graf anode of Li ion battery It succeeded for the first time in the world to conduct first principles simulation at a scale exceeding (about 2400 atoms). As a result, we discovered the effect of increasing the Li ion permeability by the salt added to the electrolyte.
Li ion batteries are currently most used mainly for electronic devices, because of its high voltage among various types of storage batteries, capable of repeated use many times and being compact. However, in order to use Li ion batteries as power for automobiles and the like, it is necessary to further improve the performance in various aspects. In the interior of the Li-ion battery, a thin film called a solid electrolyte film (SEI) is formed at the interface between the negative electrode and the electrolyte at the initial charge. Increasing the rate at which Li ions pass through the SEI is important for improving the output performance of the battery. We used the electron state calculation code DC-RGDFT developed independently to calculate 5 times the Li ion group passing through the SEI – electrolyte interface at the vicinity of the Graf anode of Li ion battery It succeeded for the first time in the world to conduct first principles simulation at a scale exceeding (about 2400 atoms). As a result, we discovered the effect of increasing the Li ion permeability by the salt added to the electrolyte.
Key words: quantum mechanics, first principles calculation, Li ion secondary battery, solid electrolyte film (SEI)
Elucidation of Ion Conduction Mechanisms in Solid Electrolytes
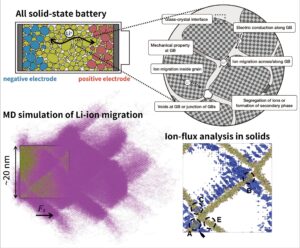 All-solid-state batteries are anticipated as the next-generation lithium-ion secondary batteries due to their advantages in safety, heat resistance, and high energy density. In all-solid-state batteries, the solid electrolyte that separates the cathode and anode is required to possess properties such as high ion conductivity, chemical stability, and resistance to lithium metal dendrites. Consequently, extensive materials research has been conducted. However, grain boundaries that separate the crystal grains in polycrystals often differ from the bulk properties, exhibiting higher ion conduction resistance, higher electronic conductivity, and lower mechanical strength. Elucidating and controlling these properties at grain boundaries are critical challenges for achieving high-performance all-solid-state batteries. Our laboratory is engaged in research to elucidate the mechanisms of ion conduction resistance at grain boundaries and heterogeneous interfaces in solid electrolytes using molecular dynamics simulations.
All-solid-state batteries are anticipated as the next-generation lithium-ion secondary batteries due to their advantages in safety, heat resistance, and high energy density. In all-solid-state batteries, the solid electrolyte that separates the cathode and anode is required to possess properties such as high ion conductivity, chemical stability, and resistance to lithium metal dendrites. Consequently, extensive materials research has been conducted. However, grain boundaries that separate the crystal grains in polycrystals often differ from the bulk properties, exhibiting higher ion conduction resistance, higher electronic conductivity, and lower mechanical strength. Elucidating and controlling these properties at grain boundaries are critical challenges for achieving high-performance all-solid-state batteries. Our laboratory is engaged in research to elucidate the mechanisms of ion conduction resistance at grain boundaries and heterogeneous interfaces in solid electrolytes using molecular dynamics simulations.
- R. KOBAYASHI, et al., Acta Materialia, **226**, 117596 (2022).
- R. Kobayashi, et al., Journal of Solid State Electrochemistry, (2024).
Key words: All-solid-state batteries, solid electrolytes, solid ionics, crystal grain boundaries
Construction of interatomic potentials
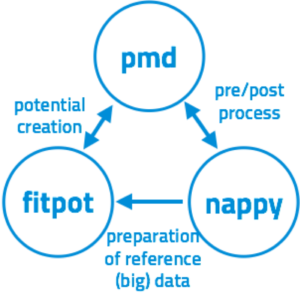 Since the molecular dynamics (MD) simulation is a very powerful tool, it is widely used in materials science. The reliability of the MD simulation strongly depends on the accuracy of the interatomic potentials used in the simulation. Interatomic potentials consist of their function forms and parameters in them. We developed a program package that can optimize the potential parameters of a variety of materials, enabling us to perform reliable MD simulation of any materials. The program can also construct neural-network (NN) potentials, which can reproduce ab-initio results in errors of a few meV/atom.
Since the molecular dynamics (MD) simulation is a very powerful tool, it is widely used in materials science. The reliability of the MD simulation strongly depends on the accuracy of the interatomic potentials used in the simulation. Interatomic potentials consist of their function forms and parameters in them. We developed a program package that can optimize the potential parameters of a variety of materials, enabling us to perform reliable MD simulation of any materials. The program can also construct neural-network (NN) potentials, which can reproduce ab-initio results in errors of a few meV/atom.
- nap — MD simulation program
- optzer — optimization of interatomic potential parameters
- R. KOBAYASHI, J. Open Source Software, **6**(57), 2768 (2021).
- R. KOBAYASHI, et al., APL Materials **8**, 081111 (2020).
Keywords: interatomic potentials, neural-network potential
Simulation of polariation structure in lead-free ferroelectrics
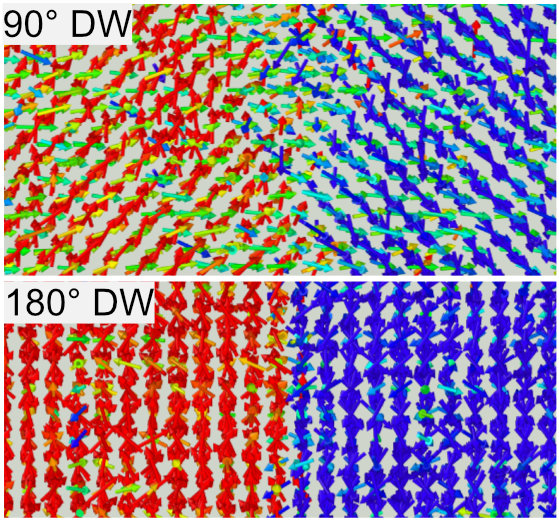 Ferroelectric materials are used in a wide range of applications such as nonvolatile memory and piezoelectric devices, taking advantage of their spontaneous polarization and piezoelectric properties. In recent years, there has been a growing interest in improving the performance of lead-free ferroelectric materials by utilizing nanostructures such as lattice defects in the materials against the backdrop of the demand for lead-free materials as a part of environmental considerations. In this laboratory, we mainly target barium titanate, a lead-free ferroelectric material, to study the effects of lattice defects on polarization inversion phenomena and the properties of polarization domain walls, which are interfaces where the direction of spontaneous polarization switches, by atomic-scale simulations. We have discovered that Ba-O and Ti-O pair defects in barium titanate significantly reduce the electric field required for polarization reversal. Furthermore, we have reproduced a 90° domain wall, a type of polarization domain wall, for the first time in finite-temperature molecular dynamics simulations for barium titanate.
Ferroelectric materials are used in a wide range of applications such as nonvolatile memory and piezoelectric devices, taking advantage of their spontaneous polarization and piezoelectric properties. In recent years, there has been a growing interest in improving the performance of lead-free ferroelectric materials by utilizing nanostructures such as lattice defects in the materials against the backdrop of the demand for lead-free materials as a part of environmental considerations. In this laboratory, we mainly target barium titanate, a lead-free ferroelectric material, to study the effects of lattice defects on polarization inversion phenomena and the properties of polarization domain walls, which are interfaces where the direction of spontaneous polarization switches, by atomic-scale simulations. We have discovered that Ba-O and Ti-O pair defects in barium titanate significantly reduce the electric field required for polarization reversal. Furthermore, we have reproduced a 90° domain wall, a type of polarization domain wall, for the first time in finite-temperature molecular dynamics simulations for barium titanate.
Keywords: molecular dynamics, ferroelectrics, lattice defect, domain wall