
ここでは我々が行なっている研究を紹介します.
( English → Please see Research page for details. )
過去の論文 → 研究成果
卒業生のテーマ → 修士論文,卒業論文
金属樹脂間の接着強度に及ぼす水分効果についてのハイブリッド量子古典シミュレーション
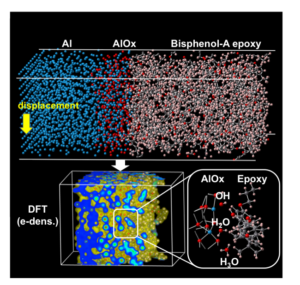 ものづくり産業を接着(密着)や封止の用途で支えているエポキシ系接着剤は,自動車のマルチマテリアル化や過酷環境での高信頼度な(圧力)センサー等の需要の高まりにより,より一層の性能向上が期待されている.本研究では,古くから知られている大問題である「水分による接着力低下現象」を,電子・原子レベルからの大規模シミュレーションにより解明することに成功した.シミュレーションに際しては,自然酸化膜を持つ金属Al系に,エポキシ樹脂系を接触させ,その接触部には水分子を導入した.また,エポキシ樹脂のモデルとして,エポキシ基の存在を強調したモデルと,OH基の存在を強調したモデルの2種類を用意した.接触部における1500原子程度の領域を,DC-RGDFTコードによるDFTで電子状態から計算し,残りの領域は古典原子間ポテンシャルにより計算した.特に,OH基の存在を強調した分子モデルを用いた場合に,実験相当の接着強度を,水分による強度低下を含めて再現することに成功した.さらに,実験測定が難しい化学反応を数種類見出した.
ものづくり産業を接着(密着)や封止の用途で支えているエポキシ系接着剤は,自動車のマルチマテリアル化や過酷環境での高信頼度な(圧力)センサー等の需要の高まりにより,より一層の性能向上が期待されている.本研究では,古くから知られている大問題である「水分による接着力低下現象」を,電子・原子レベルからの大規模シミュレーションにより解明することに成功した.シミュレーションに際しては,自然酸化膜を持つ金属Al系に,エポキシ樹脂系を接触させ,その接触部には水分子を導入した.また,エポキシ樹脂のモデルとして,エポキシ基の存在を強調したモデルと,OH基の存在を強調したモデルの2種類を用意した.接触部における1500原子程度の領域を,DC-RGDFTコードによるDFTで電子状態から計算し,残りの領域は古典原子間ポテンシャルにより計算した.特に,OH基の存在を強調した分子モデルを用いた場合に,実験相当の接着強度を,水分による強度低下を含めて再現することに成功した.さらに,実験測定が難しい化学反応を数種類見出した.
キーワード:有機無機界面,エポキシ樹脂,Al,接着,ハイブリッドシミュレーション
大規模系の電子状態計算コードの開発
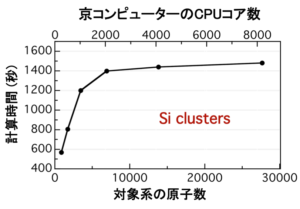 数千原子規模の大規模な対象系にも適用できる,新しい電子状態計算法とその汎用コードを研究・開発している.Kohn-Shamの定式化に基づく密度汎関数法のコードは,電子軌道を多数の平面波を使って簡便に表現し計算することがしばしば行われる.しかしその結果,スパコンの多数のノードを使った計算の際には効率が良くない.そのため,原子のダイナミクスを調べるシミュレーションでは,原子数は百オーダーが実用上の限界である.これに対して我々は,実空間での格子点上の数値として軌道関数を表現する実空間グリッド密度汎関数コード(RGDFT)を開発した.実空間表現であることから境界条件や全電荷数を自由に設定できる.この方法では,全格子点を仮想的に分割して各ノードが担当する格子点数を少なくすることで,スパコンの多数のノードを用いる際に高い性能が得られる.最近さらに,数千原子規模の対象系を扱えるように,対象系自身も仮想的にドメインに分割し,各ドメインでの実空間グリッド密度汎関数計算の結果を統合する新しい定式化も提案しコード化(DC-RGDFT)した.この方法によると,対象系の全原子に働く力を原子数に比例する計算量で得ることができる.DC-RGDFTは,スパコン京の多数のノードを用いた性能テストで,良い結果を示している.
数千原子規模の大規模な対象系にも適用できる,新しい電子状態計算法とその汎用コードを研究・開発している.Kohn-Shamの定式化に基づく密度汎関数法のコードは,電子軌道を多数の平面波を使って簡便に表現し計算することがしばしば行われる.しかしその結果,スパコンの多数のノードを使った計算の際には効率が良くない.そのため,原子のダイナミクスを調べるシミュレーションでは,原子数は百オーダーが実用上の限界である.これに対して我々は,実空間での格子点上の数値として軌道関数を表現する実空間グリッド密度汎関数コード(RGDFT)を開発した.実空間表現であることから境界条件や全電荷数を自由に設定できる.この方法では,全格子点を仮想的に分割して各ノードが担当する格子点数を少なくすることで,スパコンの多数のノードを用いる際に高い性能が得られる.最近さらに,数千原子規模の対象系を扱えるように,対象系自身も仮想的にドメインに分割し,各ドメインでの実空間グリッド密度汎関数計算の結果を統合する新しい定式化も提案しコード化(DC-RGDFT)した.この方法によると,対象系の全原子に働く力を原子数に比例する計算量で得ることができる.DC-RGDFTは,スパコン京の多数のノードを用いた性能テストで,良い結果を示している.
キーワード:量子力学,第一原理計算,スーパーコンピュータ
Liイオン二次電池の負極SEIの第一原理シミュレーション
 Liイオン電池は,様々な種類がある蓄電池の中でも電圧が高く,多数回の繰り返し利用が可能であり,コンパクトでもあるため,電子機器を中心に現在最も使われている.しかしLiイオン電池を,自動車等の動力として利用するには,さらに様々な面での性能アップが必要である.Liイオン電池の内部では,初回充電時,その負極と電解質との界面に,固体電解質皮膜(SEI)とよばれる薄い膜が生じる.LiイオンがSEIを通過する際のレートを上げることが,電池の出力性能を向上させる為に重要となっている.我々は,独自に開発した電子状態計算コードDC-RGDFTを用いて,Liイオン電池のグラファイル負極の近傍に生成されるSEI-電解液界面を透過するLiイオン群に関して,これまでの5倍を超える規模(約2400原子)での第一原理シミュレーションを行うことに世界で初めて成功した.その結果,電解質に添加する塩によるLiイオン透過率の増大効果等を発見した.
Liイオン電池は,様々な種類がある蓄電池の中でも電圧が高く,多数回の繰り返し利用が可能であり,コンパクトでもあるため,電子機器を中心に現在最も使われている.しかしLiイオン電池を,自動車等の動力として利用するには,さらに様々な面での性能アップが必要である.Liイオン電池の内部では,初回充電時,その負極と電解質との界面に,固体電解質皮膜(SEI)とよばれる薄い膜が生じる.LiイオンがSEIを通過する際のレートを上げることが,電池の出力性能を向上させる為に重要となっている.我々は,独自に開発した電子状態計算コードDC-RGDFTを用いて,Liイオン電池のグラファイル負極の近傍に生成されるSEI-電解液界面を透過するLiイオン群に関して,これまでの5倍を超える規模(約2400原子)での第一原理シミュレーションを行うことに世界で初めて成功した.その結果,電解質に添加する塩によるLiイオン透過率の増大効果等を発見した.
キーワード:量子力学,第一原理計算,Liイオン二次電池,固体電解質皮膜(SEI)
固体電解質内イオン伝導機構の解明
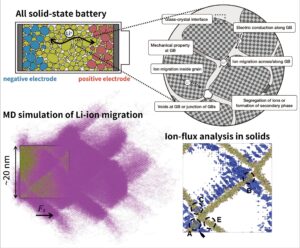 安全性,耐熱性,高エネルギー密度などの観点から,次世代Liイオン二次電池として全固体電池が期待されています.全固体電池において正極と負極を隔てる固体電解質は,高イオン伝導,化学的安定性,Li金属デンドライト耐性などの物性が要求されるため,多くの材料研究が行われています.しかしながら,多結晶体の結晶粒を隔てる粒界はバルクとは異なり,イオン伝導抵抗が高く,電子伝導度が高く,機械的強度が低いことが多いです.粒界におけるこのような物性を明らかにして,それを制御することが,高性能な全固体電池実現への課題となっています.当研究室では,分子動力学シミュレーションを用いて,固体電解質の粒界や異相界面におけるイオン伝導抵抗機構を解明する研究を行っています.
安全性,耐熱性,高エネルギー密度などの観点から,次世代Liイオン二次電池として全固体電池が期待されています.全固体電池において正極と負極を隔てる固体電解質は,高イオン伝導,化学的安定性,Li金属デンドライト耐性などの物性が要求されるため,多くの材料研究が行われています.しかしながら,多結晶体の結晶粒を隔てる粒界はバルクとは異なり,イオン伝導抵抗が高く,電子伝導度が高く,機械的強度が低いことが多いです.粒界におけるこのような物性を明らかにして,それを制御することが,高性能な全固体電池実現への課題となっています.当研究室では,分子動力学シミュレーションを用いて,固体電解質の粒界や異相界面におけるイオン伝導抵抗機構を解明する研究を行っています.
- R. KOBAYASHI, et al., Acta Materialia, **226**, 117596 (2022).
- R. Kobayashi, et al., Journal of Solid State Electrochemistry, (2024).
キーワード:全固体電池,固体電解質,固体イオニクス,結晶粒界
原子間ポテンシャル作成ツール開発
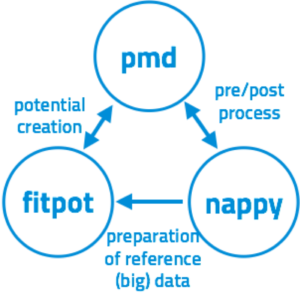 分子動力学(MD)シミュレーションは非常に強力なツールですが,そのシミュレーションが現実をよく再現しているかどうかは,原子間ポテンシャル(原子間の相互作用)の精度に強く依存します.原子間ポテンシャルはその関数形と含まれるパラメータにより決まります.本研究室では,第一原理計算を再現するようにパラメータを最適化するツールを開発しています.これを用いることで,いかなる材料であっても第一原理計算の結果を再現する原子間ポテンシャルを構築してMDシミュレーションが可能となります.また,近年活発に開発されるようになってきている機械学習型(ニューラル・ネットワーク(NN))ポテンシャルも構築することができ,これを用いることで数meV/atom精度で第一原理計算結果を再現するMDシミュレーションが可能となります.
分子動力学(MD)シミュレーションは非常に強力なツールですが,そのシミュレーションが現実をよく再現しているかどうかは,原子間ポテンシャル(原子間の相互作用)の精度に強く依存します.原子間ポテンシャルはその関数形と含まれるパラメータにより決まります.本研究室では,第一原理計算を再現するようにパラメータを最適化するツールを開発しています.これを用いることで,いかなる材料であっても第一原理計算の結果を再現する原子間ポテンシャルを構築してMDシミュレーションが可能となります.また,近年活発に開発されるようになってきている機械学習型(ニューラル・ネットワーク(NN))ポテンシャルも構築することができ,これを用いることで数meV/atom精度で第一原理計算結果を再現するMDシミュレーションが可能となります.
- nap — MDシミュレーションプログラム
- optzer — 原子間ポテンシャル・パラメータ最適化プログラム
- R. KOBAYASHI, J. Open Source Software, **6**(57), 2768 (2021).
- R. KOBAYASHI, et al., APL Materials **8**, 081111 (2020).
キーワード:原子間ポテンシャル,ニューラル・ネットワーク・ポテンシャル
非鉛系強誘電体における分極構造のシミュレーション
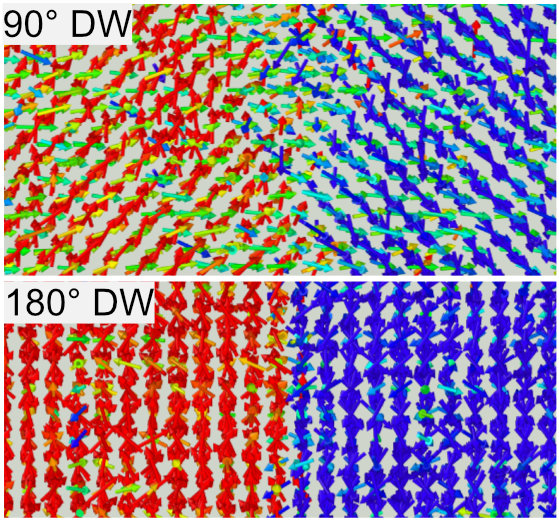 強誘電体材料は,その自発分極や圧電性を活かして,不揮発性メモリや圧電素子などの幅広い用途に利用されています.近年では,環境配慮の一環として鉛フリーの材料が望まれていることを背景に,材料内の格子欠陥などのナノ構造を活用した非鉛系強誘電体材料の性能改善が注目されています.本研究室では,非鉛系強誘電体材料の一つであるチタン酸バリウムを主なターゲットとして,格子欠陥が分極反転現象に及ぼす影響や,自発分極の向きが切り替わる界面である分極ドメイン壁の性質などを,原子スケールのシミュレーションによって研究しています.これまでに,チタン酸バリウム内のBa-OやTi-Oペア欠陥が,分極反転に必要な電場を大きく減少させることを発見しました.さらに,分極ドメイン壁の一種である90° ドメイン壁を,チタン酸バリウムに対する有限温度の分子動力学シミュレーションで初めて再現しました.
強誘電体材料は,その自発分極や圧電性を活かして,不揮発性メモリや圧電素子などの幅広い用途に利用されています.近年では,環境配慮の一環として鉛フリーの材料が望まれていることを背景に,材料内の格子欠陥などのナノ構造を活用した非鉛系強誘電体材料の性能改善が注目されています.本研究室では,非鉛系強誘電体材料の一つであるチタン酸バリウムを主なターゲットとして,格子欠陥が分極反転現象に及ぼす影響や,自発分極の向きが切り替わる界面である分極ドメイン壁の性質などを,原子スケールのシミュレーションによって研究しています.これまでに,チタン酸バリウム内のBa-OやTi-Oペア欠陥が,分極反転に必要な電場を大きく減少させることを発見しました.さらに,分極ドメイン壁の一種である90° ドメイン壁を,チタン酸バリウムに対する有限温度の分子動力学シミュレーションで初めて再現しました.
キーワード:分子動力学,強誘電体材料,格子欠陥,分極ドメイン壁